Молекулярная система смерти готовит посредников

В обзорной статье группы авторов из Института биологии развития им. Н.К. Кольцова рассматривается роль процессов деградации белков в обеспечении пластичности иммунной и нервной систем. Основой согласованного развития и функционирования этих систем являются общие для них элементы (сигнальные молекулы, трансмиттеры, рецепторы, и др.). Убиквитин-протеасомная система играет особую роль в создании и деградации молекул-посредников, т.е. регуляции биохимических процессов взаимодействия систем.
Иного рода результат регуляции убиквитин-протеасомной системой показан на примере развития иммунологической толерантности клеток печени. Поскольку организм должен быть толерантен к множеству антигенов, поступающих из желудочно-кишечного тракта, в печени сформировался специфический локальный контроль иммунного ответа. Благодаря определенному субъединичному составу и перераспределению пулов протеасом иммунные клетки перепрограммируются на «принятие» антигенов. Эти эффекты рассматриваются как перспективные для клинической трансплантологии.
Убиквитин-протеасомная система выполняет важные функции как в развивающейся и нормально функционирующей, так и в поврежденной нервной системе у разных животных. Доказано участие протеасом в процессах синаптической пластичности и долговременной пластичности в ЦНС. Деградация белков, по-видимому, играет важную роль на определенном этапе формирования памяти. Оказалось, что в нейронах и глиальных клетках млекопитающих присутствуют иммунные протеасомы. Их высокое содержание характерно для пирамидальных нейронов коры и гиппокампа. Таким образом, функционирование нервной и иммунной систем у многоклеточных организмов от насекомых до млекопитающих регулируется согласованно, с использованием общих механизмов и под контролем убиквитин-протеасомной системы.
Деградация белков-циклинов типа D

Выявлен молекулярный механизм разрушения одного из ключевых видов белков клеточного цикла — циклинов типа D. Этот механизм может лежать в основе отсутствия ответа некоторых опухолей на лечебное воздействие ингибиторными препаратами.
Белки, называемые циклинами D-типа (циклин D1, D2 и D3), — это ключевые компоненты ядерного комплекса, управляющего делением клетки. В своих работах в «Nature» Simoneschi с соавт. [1], Chaikovsky с соавт. [2] и Maiani с соавт. [3] отвечают на основные вопросы о деградации D-циклинов.
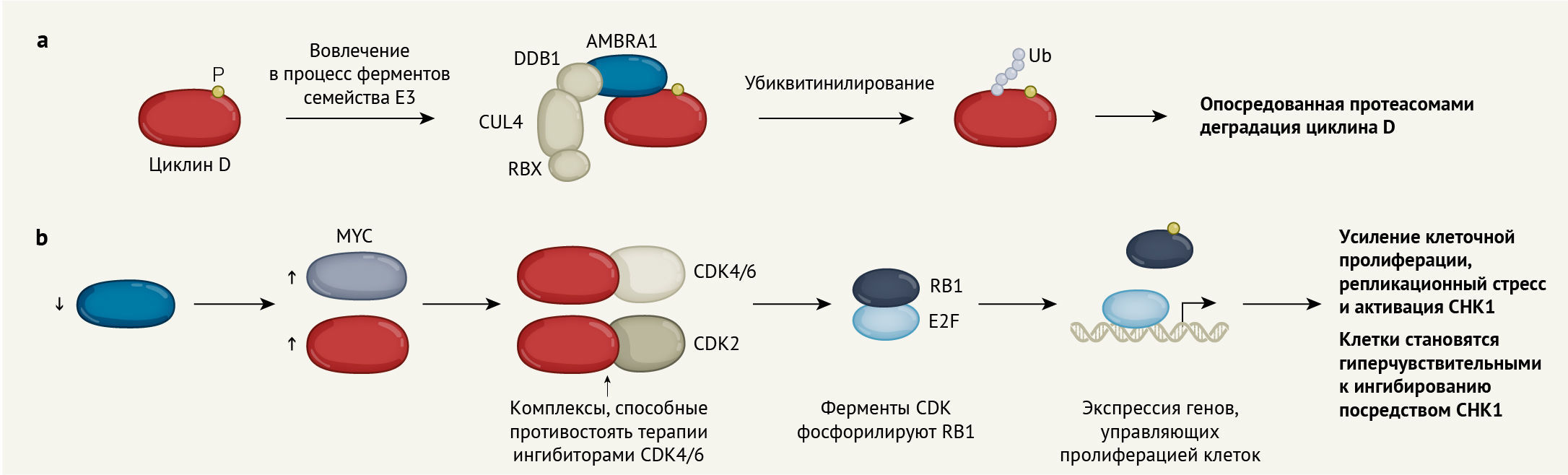
Во время деления клетки циклины D-типа связываются со своими ферментативными партнерами, называемыми циклин-зависимой киназой 4 (CDK4) и циклин-зависимой киназой 6 (CDK6), и активируют их. Эти киназы фосфорилируют белки-супрессоры опухолей RB1, RBL1 и RBL2, и это способствует клеточному делению (рис. 1). Неконтролируемая активация комплексов циклина D и киназ CDK4/6 является движущей силой в развитии многих типов рака [4].
В последнее время возрос интерес к метаболизму циклина D и CDK4/6, поскольку в клиническое применение вошли низкомолекулярные ингибиторы киназ CDK4/6. Результаты клинических испытаний этих веществ свидетельствуют об их способности увеличивать продолжительность жизни людей с раком груди. Ингибиторы CDK4/6 (палбоциклиб, рибоциклиб и абемациклиб) получили одобрение для лечения запущенных форм рака молочной железы. Более того, эти препараты задействованы в нескольких сотнях клинических испытаний, в ходе которых происходит поиск способов лечения многих различных типов рака [4, 5].
Циклины D-типа интенсивно изучаются с момента их открытия в 1990-е годы, но вопрос о том, как происходит их деградация во время клеточного цикла, остается спорным. Фосфорилирование карбоксильной концевой области D-циклинов запускает разрушение этих белков с помощью пути деградации, опосредованного убиквитин-протеасомной системой [6]. Система состоит из комплекса ферментов, включающего активатор убиквитина (E1), конъюгатор убиквитина (E2) и убиквитин-лигазу (E3); эти структуры связывают цепи нескольких молекул небольшого белка убиквитина с белками-мишенями посредством процесса, называемого убиквитинилированием. Эти убиквитинилированные белки затем подвергаются деградации в белковом комплексе, называемом протеасомой.
Самым большим семейством среди E3 являются cullin-RING лигазы (CRL). CRL состоят из cullin-белка, белка RING (который запускает E2), адаптивного белка и одного из многих различных белков субстрата-рецептора, ответственных за вовлечение целевого белка в комплекс E3 [7–9]. Некоторые субстратные рецепторы к E3, называемые CRL1, вовлечены в деградацию циклина D1, другие считаются мишенями воздействия для циклинов D2 и D3 для разрушения в протеасомах. Кроме того, как было выявлено, циклин D1 убиквитинилируется комплексом, запускаемым в анафазу, E3, который воздействует на несколько белков клеточного цикла [9]. В отличие от этих моделей функционирования, результаты других исследований [10] свидетельствуют, что на уровень и стабильность циклина D1 не влияет снижение содержания этих белков. Это указывает на то, что другие E3 регулируют деградацию циклина D1.
В статьях Simoneschi, Chaikovsky, Maiani с соавт. сообщается, что три циклина D-типа убиквитинилирутся и воздействуют на протеасомную деградацию с помощью подвида E3, называемого CRL4, с белком AMBRA1 в качестве рецептора субстрата. Уже было известно, что AMBRA1 играет ключевую роль в регуляции аутофагии — процесса, при котором клетки разрушают поврежденные органеллы или белковые структуры [11, 12]. Белок AMBRA1 также удалось идентифицировать как субстратный рецептор E3, в том числе и CRL4 [7, 13]. Проведя серию экспериментов с использованием методов клеточной, молекулярной биологии и генетики эмбриогенеза для получения сведений, одинаково применимых для биологии как мышей, так и человека, Simoneschi, Chaikovsky and Maiani с соавт. удалось продемонстрировать, что снижение концентрации AMBRA1 в нормальных и раковых клетках, а также в развивающихся эмбрионах мыши приводит к повышению содержания циклинов D-типа. Это приводит к большей степени фосфорилирования RB1 и большей пролиферации клеток, нежели чем это происходит в клетках с нормальным количеством AMBRA1.
Maiani с соавт. также продемонстрировали, что снижение содержания AMBRA1 приводит к повышению концентрации белка фактора транскрипции N-MYC. Этой же группой исследователей ранее было показано [14], что AMBRA1 регулирует стабильность и активность еще одного фактора транскрипции c-MYC. Белки семейства MYC могут повышать экспрессию циклинов D-типа и E-типа [15], тем самым ускоряя клеточный цикл.
Исходя из этих наблюдений, предполагается, что AMBRA1 может действовать как белок-супрессор опухоли. Действительно, у мышей с одной функциональной копией гена, кодирующего AMBRA1, чаще возникают опухоли легких, печени и почек, чем у нормальных мышей с двумя функциональными копиями этого гена [14]. Эти новые исследования предоставляют убедительные доказательства в поддержку данной гипотезы.
Авторы показывают, что при онкологическом заболевании у человека ген AMBRA1 мутирован. Как и ожидалось, учитывая то, что AMBRA1 способствует деградации циклина D1, авторы указывают, что уровень AMBRA1 в опухолевых тканях человека находился в обратной корреляционной зависимости от уровня циклина D1. Более того, низкий уровень AMBRA1 в опухолях оказался связан с плохим прогнозом для людей с онкологическими процессами. Экспериментальная инактивация AMBRA1 либо в опухолевых клетках человека, либо в клетках мыши, выращенных с помощью методов генной инженерии с мутациями, приводящими к развитию рака, увеличила опухолевый потенциал клеток, что было оценено после введения инъекции мышам. Кроме того, удаление гена AMBRA1 стимулировало образование опухоли (что было продемонстрировано на мышиной модели рака легкого), вызванной мутантной версией гена Kras, и у этих AMBRA1-дефицитных опухолей обнаружился более высокий уровень D-циклинов. Данные всех трех исследований свидетельствуют о том, что в основном AMBRA1 сдерживает пролиферацию клеток, не давая D-циклинам достигать высоких концентраций.
Кроме того, Chaikovsky, Simoneschi с соавт. демонстрируют, что исчезновение AMBRA1 и соответствующее повышение концентрации D-циклинов приводит к снижению чувствительности опухолевых клеток человека к ингибиторам CDK4/6. Интересно, что эти же авторы указывают, что в клетках со сниженной концентрацией AMBRA1, а не в комплексе с CDK4/6, циклин D1 также образует каталитически активный комплекс с циклин-зависимым ферментом киназой CDK2. Также у этих комплексов отсутствует чувствительность к ингибиторам CDK4/6.
Maiani с соавт. также указывают, что исчезновение AMBRA1 и, как следствие, повышение содержания D-циклинов (и, возможно, также других белков, таких как c-MYC), вызывает повреждение ДНК и репликационный стресс, который сопровождается активацией фермента киназы CHK1. Важно отметить, что Maiani с соавт. сообщают о том, что раковые клетки со сниженной концентрацией AMBRA1 гиперчувствительны к лечению ингибиторами CHK1, что предполагает потенциальную возможность терапевтического воздействия на опухоли, у которых обнаруживается дефицит AMBRA1.
Эти удивительные результаты поднимают несколько важных вопросов. Например, может ли снижение уровня AMBRA1 лежать в основе существующей или приобретенной устойчивости опухолей человека к ингибиторам CDK4/6? Является ли повышение содержания D-циклинов, которое наблюдается при исчезновении AMBRA1, единственным фактором, ответственным за развитие устойчивости к ингибиторам CDK4/6? Анализ клинических испытаний на людях с раком груди [16–19] не выявил корреляции между наличием добавочных копий гена, кодирующего циклин D1, или уровнем матричной РНК циклина-D1, или белка в опухолях и реакцией пациента на ингибиторы CDK4/6. Действительно, Chaikovsky с соавт. обнаружили, что раковые клетки человека, у которых более выражена экспрессия D-циклинов по сравнению с нормой, не обладают всеми характеристиками устойчивости к ингибиторам, как при почти полном исчезновении AMBRA1. Возможно, другие белки, регулируемые AMBRA1, такие как c-MYC, которые могут активировать белок циклин E и комплексы циклин E-CDK2, влияют на устойчивость к лечению.
Наблюдение Chaikovsky с соавт. и Simoneschi с соавт. того, как образуются комплексы циклин D – CDK2, устойчивые к препаратам-ингибиторам CDK4/6, в клетках, в которых снижено содержание AMBRA1, является невероятно занимательным для молекулярной онкологии. Как было обнаружено ранее, такие «атипичные» комплексы лежат в основе приобретенной устойчивости к ингибированию CDK4/6 [20]. Итак, можно предположить, что снижение концентрации AMBRA1 каким-то образом способствует образованию этих комплексов циклина D с CDK2, и это наряду с повышением концентрации циклина D приводит к устойчивости к ингибиторам CDK4/6. Особенно интересная перспектива, проистекающая из работы Maiani с соавт., заключается в том, что ингибиторы CHK1 можно использовать для лечения опухолей, устойчивых к ингибиторам CDK4/6, в которых крайне мало содержание AMBRA1.
Необходимы дальнейшие исследования роли AMBRA1 в развитии рака у человека. Опосредуется ли супрессивная функция AMBRA1 на опухоль преимущественно циклином D1 или c-MYC, либо же она также опосредована иными соединениями? Опухолевые клетки, которые больше не продуцируют RB1, не требовательны к наличию D-циклинов для клеточного цикла [4], поэтому, как можно предположить, исчезновение AMBRA1 будет наблюдаться в опухолях, которые продуцируют RB1, в случае, если D-циклины являются основной молекулярной мишенью AMBRA1. Также еще предстоит определить, является ли исчезновение AMBRA1 в опухолях человека взаимоисключающим параметром в связи с наличием мутаций, затрагивающих C-концы D-циклинов, которые, как предполагают, могут сделать D-циклины устойчивыми к деградации, опосредованной AMBRA1. Другой нерешенный вопрос заключается в том, почему (по данным Chaikovsky с соавт.), существует корреляционная зависимость между низкой степенью экспрессии AMBRA1 и высокими концентрациями циклина D1. Также неясно, почему эта низкая степень экспрессии AMBRA1 связана с плохой выживаемостью при опухолях легких, в которых присутствуют определенные типы генетических изменений, например, мутации Kras. Этот эффект не наблюдается для опухолей легких, в которых вместо этой мутации были мутантные версии гена, кодирующего белок EGFR, или Kras дикого типа.
Независимо от ответов на эти вопросы, впечатляющие исследования Chaikovsky с соавт., Maiani с соавт. and Simoneschi с соавт. вносят свой важный вклад в понимание механизмов, управляющих клеточным циклом.
Что такое деградация белка
• Транслоцированные в ЭПР белки могут экспортироваться обратно в цитозоль, где они подвергаются убиквитинилированию и деградации протеосомами — процесс известный как деградация, связанная с ЭПР.
• Белки возвращаются в цитозоль путем ретроградной транслокации, механизм которой менее понятен, чем механизм транслокации в ЭПР.
Контроль за структурой белков, осуществляемый в ЭПР, обеспечивает экспорт только таких трансмембранных и секреторных белков, которые обладают правильной нативной конфигурацией, в то время как белки неправильной структуры удерживаются в ЭПР шаперонами и имеют шанс исправить конфигурацию. Однако что же происходит с белками, которым не удается приобрести правильную структуру?
Такие белки должны каким-то образом деградировать. Одним из путей решения этой проблемы служит использование ретроградной транслокации (иногда называемой дислокация или ретротранслокация) для экспорта неправильно собранных белков обратно в цитозоль. Там они взаимодействуют с убиквитином и деградируют с участием больших протеазных комплексов, протеосом. Этот путь деградации называется — деградация, связанная с ЭПР (ERAD).
Открытие этого пути последовало после понимания того факта, что белки, обладающие неправильной нативной структурой, не накапливаются в ЭПР, а деградируют. Первоначально предполагалось, что деградация происходит в самом ЭПР, однако, несмотря на тщательные поиски, в этой органелле не были обнаружены соответствующие протеазы. Вместо этого начали появляться данные о том, что белки деградируют под действием протеаз, содержащихся в цитозоле.
Вначале было показано, что химические ингибиторы протеосом блокируют деградацию вновь синтезированного интегрального мембранного белка (CFTR, или регулятор трансмембранного переноса при кистозном фиброзе). Эти данные позволили предположить, что белки должны экспортироваться из ЭПР до деградации. В дальнейшем было показано, что ингибирование функций протеосом приводит к накоплению в цитозоле полностью транслоцированных белков.
Каким образом белок отбирается для ERAD? В отличие от процесса транслокации в ЭПР сигнальная последовательность на белке для его направления на деградацию отсутствует. Скорее всего, при ERAD распознаются какие-то свойства, которые отличают белки с неправильной структурой от белков, обладающих нативной конформацией или временно не имеющих правильной структуры высшего порядка.
Например, мутация, которая дестабилизирует нативную структуру белка, может вызывать ее деградацию. Фактически мутировавший белок может быть признан субстратом для ERAD, даже если мутация мало повлияла на его функцию. Даже белки, не обладающие заметными дефектами структуры, могут быть отобраны для ERAD, что свидетельствует о существовании скрытых, неизвестных нам признаков, которые, однако, узнаются клеткой.
Рисунок ниже иллюстрирует основные этапы ERAD. Первый этап отбора белка для ERAD — это обнаружение неправильной структуры. Хотя мы и не знаем, каким образом это происходит, возможно, что существенную роль здесь играют шапероны. Однако ассоциация с шаперонами не может являться единственным критерием отбора белка для ERAD, поскольку шапероны участвуют в организации конформации всех новообразующихся белков. Не исключено, что сигнал к ретроградной транслокации зависит от промежутка времени, в течение которого белок находится в связанной с шаперонами форме.
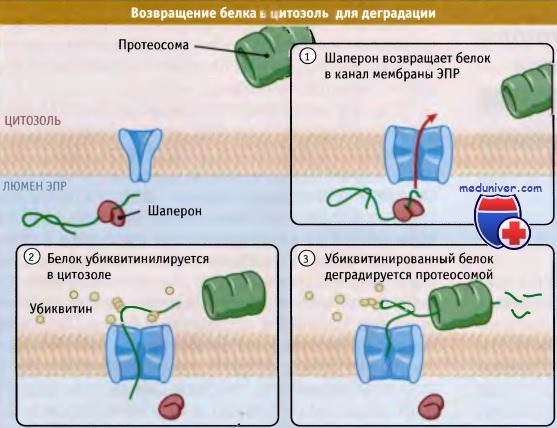 При ретроградной транслокации белки с неправильной нативной структурой возвращаются из люмена эндоплазматического ретикулума (ЭПР) обратно в его мембрану и в цитоплазму.
При ретроградной транслокации белки с неправильной нативной структурой возвращаются из люмена эндоплазматического ретикулума (ЭПР) обратно в его мембрану и в цитоплазму.
Там они подвергаются деградации в протеосомах. По-видимому, определенную роль в переадресовании играют шапероны люмена,
поскольку они должны связываться с белками, обладающими неправильной нативной структурой.
Шапероны остаются в связанной форме до тех пор, пока белок не приобретет нативную структуру. Если образование такой структуры невозможно, то белок дольше, чем обычно, будет оставаться связанным с шаперонами. Впрочем, вообще не ясно, каким образом продолжительность связи белка с шаперонами дает сигнал к ретроградной транслокации. В случае неправильной структуры гликопротеинов фермент а-маннозидаза I отщепляет маннозу и тем самым дает сигнал к деградации белка. Не ясно, однако, что служит сигналом к отщеплению маннозы.
После отбора белка для ретроградной транслокации, он должен пройти через мембрану ЭПР. Многочисленные генетические и биохимические данные позволяют предполагать, что роль канала ретроградной транслокации выполняет комплекс Sec61p. В некоторых случаях, однако, ретроградная транслокация не зависит от Sec61, и поэтому возникает вопрос о природе канала или даже о наличии нескольких каналов, специфичных для различных типов белков, обладающих неправильной структурой. Независимо от природы канала, для ретроградной транслокации надо выполнять те же шаги, как и для прямой транслокации.
Для того чтобы прошел экспорт, белок должен вначале попасть в канал, канал должен открыться, и затем — белок пройти через него в цитозоль.
Большинство белков направляются на деградацию только после завершения транслокации. Неизвестно, остаются ли эти белки поблизости от транслокона, пока не разрешится вопрос об их деградации. Также неизвестно, каким образом неправильно скрученные белки попадают в канал ретроградной транслокации. Фактически вырисовывающаяся картина позволяет предполагать, что путь, по которому направляется субстрат из ЭПР, и белки цитозоля, необходимые для деградации, сильно зависят от типа деградируемого белка и от того, где в нем располагается неправильно скрученная область.
Не исключено, что растворимые и трансмембранные белки деградируют по крайней мере частично отличающимся друг от друга путями и что среди трансмембранных белков эти пути зависят от положения мутации в домене люмена или цитозоля.
Как только белок оказался в ретроградном транслоконе, он, вероятно, начинает проходить через канал под действием сил цитозоля. Убиквитинилирование играет роль в экспорте многих (хотя и не всех) субстратов. Большинство субстратов деградации подвергается полиубиквитинилированию, и убиквитинилирование происходит, пока они еще находятся в связанном с мембраной состоянии. Мутации в каком-либо компоненте системы убиквитина приводят к образованию в ЭПР агрегатов, состоящих из белков неправильной структуры. Это позволяет предполагать, что канал блокируется в случае, когда субстрат не может модифицироваться.
Однако одного убиквитинилирования недостаточно для перехода белков в цитозоль. Также необходимо присутствие АТФазы цитозоля (р97 Cdc48 у дрожжей), которая связывается с мембраной ЭПР, при этом р97 связывает цитозольные кофакторы, которые прямо соединяются с субстратом ERAD. Пока неясно, каким образом эта АТФаза приводит к высвобождению субстрата в цитозоль, но аналогичные АТФазы бактерий и митохондрий непосредственно связываются с интегральными мембранными белками и извлекают их из мембраны. Не исключено, что АТФаза, требующаяся для ретротранслокации, действует аналогичным образом, связываясь или непосредственно с субстратом, или с присоединенным к нему остатком убиквитина и вытягивая белок из канала в цитозоль. Пока неизвестно, какую роль играет протеосома в процессе ретроградной транслокации.
Еще одним ключевым компонентом системы ERAD является небольшой мембранный белок дерлин. Этот белок необходим для ретроградной транслокации некоторых белков, обладающих неправильной структурой. Показано, что он взаимодействует как с неправильно скрученным белком в ЭПР, так и с р97 через связывание с белком VIMP. Поэтому дерлин представляет собой, по крайней мере, один компонент молекулярного «моста» между люменом ЭПР и аппаратом деградации цитозоля, хотя, конечно, существуют и другие.
Как иллюстрирует рисунок ниже, в некоторых случаях белки могут отбираться для деградации даже до завершения их транслокации в ЭПР. Это используется для особенно больших субстратов для того, чтобы избежать затрат энергии на транслокацию белка целиком, если он начинает принимать неправильную конфигурацию уже в самом начале синтеза. Отбор белков для ретроградной транслокации в процессе их импорта позволяет не помечать их повторно. Наилучшим примером этому является деградация аполипопротеина В. Это очень большой секретируемый белок, который на первом шаге сборки липопротеинов низкой плотности связывается с липидами люмена ЭПР и жирными кислотами.
Неспособность фермента люмена ЭПР переносить липиды на аполипопротеин В по мере транслокации вызывает начало деградации белка до момента завершения его синтеза и транслокации.
Когда белки достигают цитозоля, они обычно узнаются и деградируют. Однако в некоторых случаях экспортируемые белки подвергаются агрегации в цитоплазме. При этом белки накапливаются в цитоплазме в специальных образованиях, которые называются агресомы. Они способны сохранять для деградации большие количества белка.
Исследование ERAD еще только начинается. Очевидно, в этом процессе участвует больше компонентов, чем известно на сегодняшний день, и его молекулярные основы исследованы пока недостаточно. Сюда относятся фундаментальные вопросы, связанные с распознаванием белков, обладающих неправильной структурой. Выяснение деталей этих процессов позволит реконструировать их in vitro.
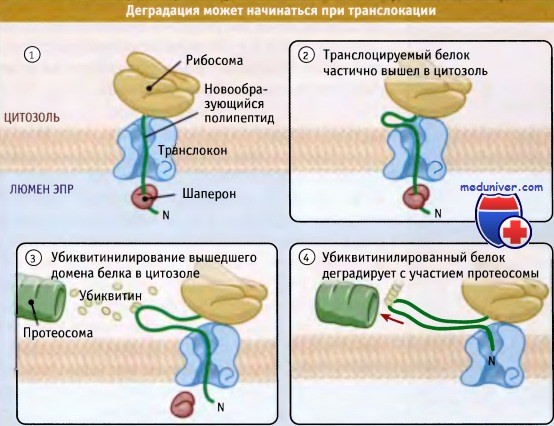 Деградация некоторых белков начинается до того момента, как они полностью транслоцировались.
Деградация некоторых белков начинается до того момента, как они полностью транслоцировались.
Когда белок отобран для деградации, в цитоплазме начинают накапливаться и убиквитироваться фрагменты его цепи.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Медицинские интернет-конференции
Языки
Апоптоз: молекулярно-клеточные механизмы развития, значение в обеспечении клеточного гомеостаза.
Булудова М.В., Полутов В.Э.
Резюме
В работе представлены современные данные литературы, относительно механизмов развития апоптоза, его принципиальных отличий от некроза, значение в поддержании клеточного гомеостаза в лимфоидной и других пролиферирующих тканях.
Ключевые слова
Статья
Апоптоз — форма гибели клетки, проявляющаяся в уменьшении ее размера, конденсации и фрагментации хроматина, уплотнении цитоплазматической мембраны без выхода содержимого клетки в окружающую среду. (Попков В.М., Чеснокова Н.П., Барсуков В.Ю., 2011).
Апоптоз играет жизненно важную роль в процессе эмбрионального и онтогенетического развития, имеет место при различных морфогенетических процессах, обеспечивает поддержание клеточного гомеостаза как в лимофоидной ткани, так и в других пролиферирующих тканях. Нарушение апоптоза в эмбриогенезе может приводить к внутриутробной гибели плода, врожденным уродствам или различным заболеваниям, в том числе и злокачественным новообразованиям.
а) переход фосфатидилсерина из внутреннего монослоя цитоплазматической мембраны в наружный монослой; выход цитохрома С из межмембранного пространства митохондрий в цитоплазму
б) активация цистеиновых протеиназ (каспаз)
в) образование активных форм кислорода
г) сморщивание (blebbing) цитоплазматической мембраны
д) последующий распад ядра на части
е) фрагментация клеток на везикулы с внутриклеточным содержимым — апоптотические тельца
ж) апоптотические тела захватываются фагоцитирующими клетками микроокружения, как в случае некроза. При развитии апоптоза выброса клеточного содержимого не происходит, воспаление не возникает. Некроз распространяется обычно на группы клеток, в то время как апоптоз носит селективный характер в отношении отдельных клеток (Дмитриева Л.А., Максимовский Ю.М., 2009).
Стадии апоптоза
Стадия инициации. На этой стадии патогенный агент либо сам является информационным сигналом, либо обусловливает генерацию сигнала в клетке и его проведение к внутриклеточным регуляторным структурам и молекулам. Инициирующие апоптоз стимулы могут быть трансмембранными или внутриклеточными. Трансмембранные сигналы подразделяют на отрицательные и положительные. Отрицательные сигналы обусловливают отсутствие или прекращение воздействия на клетку различных факторов роста, регулирующих деление и созревание клетки. Положительные сигналы генерируют запуск программы апоптоза. Так, связывание TNFα (FasL) с его мембранным рецептором CD95 (Fas) активирует программу смерти клетки. Среди внутриклеточных стимулов апоптоза зарегистрированы избыток Н+, свободные радикалы липидов и других веществ, повышенная температура, внутриклеточные вирусы и гормоны, реализующие свой эффект через ядерные рецепторы (например, глюкокортикоиды).
Стадия удаления фрагментов погибших клеток. На поверхности апоптозных телец экспрессируются лиганды, с которыми взаимодействуют рецепторы фагоцитирующих клеток. Фагоциты быстро обнаруживают, поглощают и разрушают апоптозные тельца. Благодаря этому содержимое разрушенной клетки не попадает в межклеточное пространство. (Лихтенштейн А.В., Шапот В.С., 1998).
Одно из апоптотических событий реализуется в ядре клетки и заключается в фрагментации ДНК. Деградация ДНК является терминальной фазой апоптоза, связанной с проявлением активности различных эндонуклеаз, последние обусловливают либо появление крупных фрагментов ДНК, или развитие межнуклеосомальной деградации ДНК. Считают, что этот тип деградации обеспечивается активацией Са2+, Mg2+-зависимой эндонуклеазы.
Исследования последних лет привели к формированию принципиально новых представлений о механизме гибели клеток, имеющих повреждения ДНК, как о процессе, осуществляемом в соответствии с определенной генетической программой. В индукции этой программы при наличии повреждений в ДНК клетки важная роль принадлежит белку р53. Этот белок с молекулярной массой 53 кД, локализован в ядре клетки и является одним из транскрипционных факторов. Повышенная экспрессия этого белка приводит к репрессии ряда генов, регулирующих транскрипцию и причастных к задержке клеток в фазе клеточного цикла G1. Если же активность репарационных систем недостаточна и повреждения ДНК сохраняются, то в таких клетках индуцируется программируемая клеточная гибель, или апоптоз, что приводит к защите организма от присутствия клеток с поврежденной ДНК, т.е. мутантных и способных к злокачественной трансформации.
Таким образом, при действии генотоксических агентов р53 не только увеличивает время репарации ДНК. но также защищает организм от клеток с опасными мутациями. (Паукова В.С., Пальцева М.А., Улумбекова Э.Г., 2015).
Механизмы индукции апоптоза
Регуляция апоптоза обеспечивается гормонами, цитокинами и в значительной мере особенностями генома. Ослабление или устранение гормональных влияний на клетки- мишени приводит, как правило, к индукции апоптоза.
Таким образом, апоптоз является тем механизмом, который обуславливает элиминацию клеток с определенной специфичностью рецепторов. (Миронова С.П., Котельников Г.П., 2013).
В настоящее время складывается впечатление о центральной роли протеаз в запуске и развитии процесса апоптоза. Причем, по-видимому, при апоптозе, в отличие от физиологического ответа клетки, действуют свои, характерные только для апоптоза, специализированные необратимые реакции протеолиза, катализируемые специфическими протеазами, относящихся к классу цистеиновых протеаз.
Роль наследственных факторов в регуляции апоптоза
Выяснение роли белков семейства Вс1-2 занимает центральное место в изучении регуляции процесса апоптоза. К настоящему времени известно, что белки этого семейства относятся либо к индукторам апоптоза (Bad, Bax, Bcl-Xs, Bik, Bid, Bak), либо к ингибиторам (Bcl-2, Bcl-XL). Белки семейства Bcl-2 находятся в постоянном динамическом равновесии, образуя гомо- и гетеродимеры, что в конечном счете влияет на развитие апоптоза клеток. Поэтому считается, что соотношение активных форм этих белков определяют реостат жизни и смерти клетки. (Попков В.М., Чеснокова Н.П., Захарова Н.Б., 2016).



