Миелодиспластические синдромы (МДС)
Миелодиспластические синдромы (МДС) – разнородная группа хронических заболеваний крови, при которых происходит нарушение созревания клеток крови, с возможным переходом в лейкоз.
Ранее МДС имел различные названия (малопроцентный острый лейкоз, предлейкоз, тлеющая лейкемия и др.).
Заболеваемость МДС составляет 3-5 случаев на 100 тысяч населения в год. Более 85% пациентов старше 60 лет, причем риск развития заболевания с возрастом увеличивается. В 2/3 случаев МДС носит вторичный характер и развивается на фоне других заболеваний крови (множественная миелома, лимфогранулематоз, неходжкинские лимфомы, хронический лимфолейкоз) и после цитостатической терапии при ревматоидном артрите, системной красной волчанке, хроническом гломерулонефрите.
Причина заболевания окончательно не выяснена, определенную роль играют ионизирующее излучение, прием цитостатические препараты, производные бензола.
ДИАГНОСТИКА
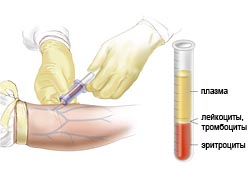
Клинический анализ крови позволяет выявить анемию (малокровие), которая является самым частым симптомом МДС и обнаруживается в 85-90% случаев, лейкопению (снижение числа лейкоцитов), выявляемую у 50% больных и тромбоцитопению (снижение количества тромбоцитов), проводящую к кровотечениям.
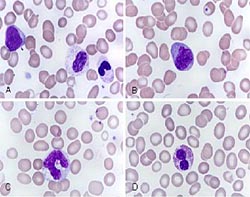

Цитогенетический анализ позволяет определить изменения в хромосомном аппарате клеток крови и является ключевым методом диагностики и определения прогноза заболевания.
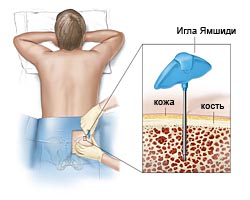
Аспирация (пункция костного мозга) и биопсия (трепанобиопсия) костного мозга – получение небольшого количества (0,2-0,5 мл) аспирата костного мозга и небольшого фрагмент кости. Процедура абсолютно безопасна и выполняется под местной анестезии в течение 2-5 мин. Может также проводиться амбулаторно. Исследования материала биопсий позволяют выявить изменения в костном мозге характерные для МДС.
У 20% пациентов можно выявить увеличение размеров селезенки, печени.
Выбор лечения определяется вариантом заболевания, прогнозом а также наличием сопутствующих заболеваний, которые могут существенно повлиять на терапию.

При благоприятном прогнозе и минимальных проявлениях заболевания можно ограничиться наблюдением за больным, пока показатели крови и костного мозга остаются стабильными.
При выраженном малокровии (анемии) показано переливание эритроцитарной массы, а при повышенной кровоточивости переливание тромбомассы. В ряде случаев возможно применение ростовых факторов (колониестимулирующие факторы).
Единственным методом лечения, позволяющим существенно увеличить продолжительность жизни больных МДС, является аллогенная трансплантация костного мозга/периферических стволовых клеток, Однако применение аллогенной трансплантации не всегда возможно в связи с пожилым возрастом большинства больных и отсутствием идентичного родственного донора.
До настоящего времени результаты лечения больных МДС остаются неудовлетворительными, в связи с этим не существует общепринятых стандартов лечения, а определены лишь общие подходы к терапии для разных групп больных при разных вариантах заболевания. Поэтому сохранение качества жизни пациентов с МДС в большинстве случаев, выходит на первый план при проведении лечения.
Синдром мышечной дистонии
Синдромом мышечной дистонии называют состояние, при котором непроизвольно сокращаются мышцы, вследствие чего нарушено естественное положение тела или его отдельных конечностей. Диагноз «синдром мышечной дистонии» и «нарушенный мышечный тонус» довольно часто ставят новорожденным детям, из-за этого у родителей возникает вопрос: насколько опасно это заболевание, и каким образом его лечить.
Причины синдрома мышечной дистонии
Виды синдрома мышечной дистонии
Синдром мышечной дистонии сопровождается незначительными отклонениями или чрезмерной скованностью отдельных частей тела и ограниченной двигательной активностью, отмечается неравномерный или переменный тонус. Специалисты могут определить гипертонус, при котором отмечается усиленное напряжение мышц, или гипотонус при пониженном напряжении мышц. Заболевание может охватить группу мышц, одну конечность (при локальной дистонии) или все тело (при диффузной или генерализованной торсионной мышечной дистонии).
Самые распространенные виды заболевания — спазматическая дисфония (при спазмах голосовых мышц), блефароспазм (при дистонии глазных мышц), кривошея (при дистонии мышц шеи) и писчий спазм (при судорогах мышц рук). Деформирующая мышечная дистония – самая серьезная форма заболевания, которая может привести к нарушениям функций организма.
Симптомы синдрома мышечной дистонии
Синдром мышечной дистонии у детей носит односторонний или ассиметричный характер, при котором малыш поворачивает голову и делает перевороты только на одну сторону – это должно обеспокоить родителей.
К главному признаку синдрома мышечной дистонии можно отнести неестественную походку и аномальные позы тела или конечностей (например, при выгибании и выкручивании).
Заболевание характеризуется нарушением двигательных навыков, происходит торможение статико-моторного развития малыша (способность ползать, сидеть, ходить). Прогрессирующий синдром мышечной дистонии ограничивает трудоспособность пациента и является причиной инвалидности, поэтому при начальных симптомах необходимо обращаться к врачу. При пониженном тонусе ребенок вялый, неактивный, мало двигается. Активность движений ребенка зависит от степени понижения тонуса скелетной мускулатуры.
Диагностика синдрома мышечной дистонии
Диагностические критерии для диагностики синдрома мышечной дистонии заключаются в следующем:
Лечение синдрома мышечной дистонии
Лечение синдрома мышечной дистонии должно быть комплексным. Оно может включать в себя массаж, физиопроцедуры, лечебную гимнастику и, если необходимо, прием медикаментозных препаратов.
С помощью определенных приемов массажист должен расслабить или привести в тонус необходимую группу мышц. Также возможно использование специальных мануальных приемов, чтобы нормализовать общее психомоторное состояние малыша.
Профилактика синдрома мышечной дистонии
Профилактикой дистонии является здоровая мама, контроль развития беременности и состояния плода, исключение вредных привычек, прохождение курса оздоровительного массажа и гимнастики, развивающий уход за малышом, комплексные осмотры для быстрой диагностики и консультация невролога.
Рабдомиолиз
, MD, University of California, San Francisco
Патофизиология рабдомиолиза
Этиология рабдомиолиза
Теоретически любая форма повреждения мышц может привести к рабдомиолизу. К наиболее распространенным причинам относятся
Менее распространенные причины включают следующее:
Чрезмерная физическая нагрузка или длительная иммобилизация
Симптомы и признаки рабдомиолиза
Классическая триада симптомов при рабдомиолизе – это мышечная боль, слабость и красновато-коричневая моча. Однако эта триада симптомов присутствует менее чем у 10% всех пациентов с рабдомиолизом.
Другие признаки и симптомы варьируются в зависимости от причины, вызвавшей заболевание, и наличия осложнений (например, у пациентов с инфекцией повышается температура, а интоксикация может вызвать нарушение психического статуса).
Справочные материалы по симптоматике
Gabow P, Kaehny W, Kelleher S: The spectrum of rhabdomyolysis. Medicine 62:141-152, 1982. doi: 10.1097/00005792-198205000-00002
Melli G, Chaudhry V, Cornblath DR: Rhabdomyolysis: An evaluation of 475 hospitalized patients. Medicine (Baltimore) 84(6):377, 2005. doi: 10.1097/01.md.0000188565.48918.41
Veenstra J, Smit WM, Krediet RT, et al: Relationship between elevated creatine phosphokinase and the clinical spectrum of rhabdomyolysis. Nephrol Dial Transplant 9(6):637, 1994. doi: 10.1093/ndt/9.6.637
Диагностика рабдомиолиза
Уровень сывороточной креатинкиназы (КК) более чем в 5 раза превышает верхний предел нормы
Подозрение на рабдомиолиз основывается на анамнезе, клинических признаках и симптомах. Подтверждение осуществляется при помощи лабораторного оценивания повышенной КК. Хотя пороговое значение не установлено, для диагностики обычно требуется уровень креатинкиназы, более чем в 5 раз превышающий верхнюю границу нормы.
Лечение рабдомиолиза
Лечение основного заболевания
В целом, лечение является поддерживающим, наряду с этиологическим лечением и коррекцией любых возникающих осложнений.
Основные положения, касающиеся рабдомиолиза
Классическая триада симптомов (миалгия, мышечная слабость и моча цвета чая) присутствует в
Обычно для диагностики требуется уровень креатинкиназы > 5 раз выше верхнего предела нормы.
Лечение состоит из поддерживающей инфузии внутривенными растворами, а также устранения причины заболевания и любых последующих осложнений.
Миелодиспластический синдром
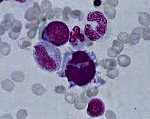
Миелодиспластический синдром – группа гематологических заболеваний, при которых наблюдаются цитопения, диспластические изменения костного мозга и высокий риск возникновения острого лейкоза. Характерные симптомы отсутствуют, выявляются признаки анемии, нейтропении и тромбоцитопении. Диагноз устанавливается с учетом данных лабораторных анализов: полного анализа периферической крови, гистологического и цитологического исследования биоптата и аспирата костного мозга и т. д. Дифференциальный диагноз может представлять значительные затруднения. Лечение – переливание компонентов крови, химиотерапия, иммуносупрессивная терапия, пересадка костного мозга.
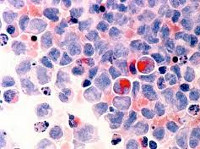
Общие сведения
Миелодиспластический синдром – группа заболеваний и состояний с нарушениями миелоидного кроветворения и высоким риском развития острого лейкоза. Вероятность развития увеличивается с возрастом, в 80% случаев данный синдром диагностируется у людей старше 60 лет. Мужчины страдают несколько чаще женщин. У детей миелодиспластический синдром практически не встречается. В последние десятилетия гематологи отмечают увеличение заболеваемости среди лиц трудоспособного возраста. Предполагается, что причиной «омоложения» болезни могло стать существенное ухудшение экологической обстановки.
До недавнего времени лечение миелодиспластического синдрома было только симптоматическим. Сегодня специалисты разрабатывают новые методы терапии, однако эффективное лечение этой группы болезней все еще остается одной из самых сложных проблем современной гематологии. Пока прогноз при миелодиспластическом синдроме, в основном, зависит от особенностей течения болезни, наличия или отсутствия осложнений. Лечение осуществляют специалисты в сфере онкологии и гематологии.
Причины и классификация миелодиспластического синдрома
С учетом причин развития различают два типа миелодиспластического синдрома: первичный (идиопатический) и вторичный. Идиопатический вариант выявляется в 80-90% случаев, диагностируется преимущественно у пациентов старше 60 лет. Причины возникновения установить не удается. В числе факторов риска первичного миелодиспластического синдрома – курение, повышенный уровень радиации при выполнении профессиональных обязанностей или проживании в неблагоприятной экологической зоне, частый контакт с бензином, пестицидами и органическими растворителям, некоторые наследственные и врожденные заболевания (нейрофиброматоз, анемия Фанкони, синдром Дауна).
Вторичный вариант миелодиспластического синдрома наблюдается в 10-20% случаев, может возникать в любом возрасте. Причиной развития становится химиотерапия или радиотерапия по поводу какого-то онкологического заболевания. В число лекарственных средств с доказанной способностью вызывать миелодиспластический синдром включают циклофосфан, подофиллотоксины, антрациклины (доксорубицин) и ингибиторы топоизомеразы (иринотекан, топотекан). Вторичный вариант отличается более высокой резистентностью к лечению, более высоким риском развития острого лейкоза и более неблагоприятным прогнозом.
В современной редакции классификации ВОЗ различают следующие типы миелодиспластического синдрома:
Симптомы миелодиспластического синдрома
Клиническая симптоматика определяется степенью нарушений миелопоэза. При мягко протекающих расстройствах возможно длительное бессимптомное или стертое течение. Из-за слабой выраженности клинических проявлений некоторые больные не обращаются к врачам, и миелодиспластический синдром обнаруживается во время проведения очередного медицинского осмотра. При преобладании анемии наблюдаются слабость, одышка, плохая переносимость физических нагрузок, бледность кожных покровов, головокружения и обморочные состояния.
При миелодиспластическом синдроме с тромбоцитопенией возникает повышенная кровоточивость, отмечаются десневые и носовые кровотечения, на коже появляются петехии. Возможны подкожные кровоизлияния и меноррагии. Миелодиспластический синдром с выраженными нейтропенией и агранулоцитозом проявляется частыми простудами, стоматитом, синуситом или стрептодермией. В тяжелых случаях возможно развитие пневмонии или сепсиса. Инфекционные заболевания нередко вызываются грибками, вирусами или условно-патогенными микробами. У каждого пятого пациента с миелодиспластическим синдромом выявляется увеличение лимфоузлов, селезенки и печени.
Диагностика миелодиспластического синдрома
При исследовании костного мозга количество клеток обычно нормальное либо повышенное. Уже на ранних стадиях обнаруживаются признаки дизэритропоэза. Количество бластов зависит от формы миелодиспластического синдрома, может быть нормальным либо увеличенным. В последующем наблюдаются дисгранулоцитопоэз и дисмегакариоцитопоэз. У некоторых больных признаки дисплазии костного мозга выражены очень слабо. В процессе цитогенетического исследования у ¾ больных выявляются хромосомные нарушения. Дифференциальный диагноз миелодиспластического синдрома проводят с В12-дефицитной анемией, фолиево-дефицитной анемией, апластической анемией, острым миелолейкозом и другими острыми лейкозами.
Лечение и прогноз при миелодиспластическом синдроме
Тактика лечения определяется выраженностью клинической симптоматики и лабораторных изменений. При отсутствии явных признаков анемии, геморрагического синдрома и инфекционных осложнений осуществляется наблюдение. При миелодиспластическом синдроме с выраженной анемией, тромбоцитопенией и нейтропенией, а также при высоком риске возникновения острого лейкоза назначают сопроводительную терапию, химиотерапию и иммуносупрессивную терапию. При необходимости осуществляют пересадку костного мозга.
Сопроводительная терапия является самым распространенным методом лечения миелодиспластического синдрома. Предусматривает внутривенные инфузии компонентов крови. При длительном применении может провоцировать повышение уровня железа, влекущее за собой нарушения деятельности жизненно важных органов, поэтому переливания гемокомпонентов производят при одновременном приеме хелаторов (лекарственных средств, связывающих железо и способствующих его выведению).
Иммуносупрессоры эффективны при лечении миелодиспластического синдрома с отсутствием хромосомных аномалий, наличием гена HLA-DR15 и гипоклеточном костном мозге. Химиотерапию применяют при невозможности трансплантации костного мозга. Высокие дозы препаратов используют при трансформации миелодиспластического синдрома в острый лейкоз, а также при рефрактерных анемиях с избытком бластов при нормоклеточном и гиперклеточном костном мозге, низкие – при невозможности пересадки костного мозга. Наряду с перечисленными средствами пациентам назначают гипометилирующие средства (азацитидин). Наиболее надежным способом достижения полноценной длительной ремиссии является трансплантация костного мозга.
Прогноз зависит от типа миелодиспластического синдрома, количества хромосомных аномалий, необходимости в регулярных переливаниях компонентов крови, выраженности клинических проявлений и наличия осложнений. Различают 5 групп риска. Средняя выживаемость больных миелодиспластическим синдромом, входящих в группу с самым низким уровнем риска, составляет более 11 лет; с самым высоким – около 8 месяцев. Вероятность отторжения костного мозга после трансплантации – около 10%.
Мочекислый диатез — всегда ли это проявление нарушенного пуринового обмена?
.gif) |
| Рис. 1. Причины мочекислой кристаллурии |
Таким образом, рассматривая мочекислый диатез с позиций сегодняшнего дня, следует отличать диатез, обусловленный особенностью пуринового обмена, от диатеза, вызванного нарушенной функцией аммониогенеза. При этом важно выделять первичную форму нарушенного аммониогенеза, которая, скорее всего, имеет семейный характер, и именно эта форма и должна характеризовать этот вид мочекислого диатеза, который в отличие от нервно-артритического диатеза целесообразно назвать МКД. Выделение этой разновидности диатеза позволит более рационально осуществлять профилактику возможного перехода данного вида диатеза в мочекислую нефропатию, а далее — в интерстициальный нефрит дизметаболического генеза или мочекаменную болезнь (рис. 2). При этом диатезе нет никакой необходимости, в отличие от нервно-артритического диатеза, строго придерживаться диеты с ограничением продуктов, богатых пуринами, ибо здесь нет нарушения пуринового обмена, а поэтому нет гиперурикемии и гиперурикозурии. Необходимо лишь поддерживать достаточный диурез и способствовать подщелачиванию мочи периодическим назначением цитратов, соответствующих минеральных вод и ряда других препаратов, стремясь поддерживать рН мочи в пределах 6,4–6,8.
.gif) |



